“parse_entrezgene.py” and “calculate_sequences.py”
are two useful script files for parsing NCBI Gene files and
NCBI Nucleotide sequences to create the GeneBase
database.
Requirements: any operating system (Linux, Mac OS X, Windows, ...) with Python 2.7 and IDLE.
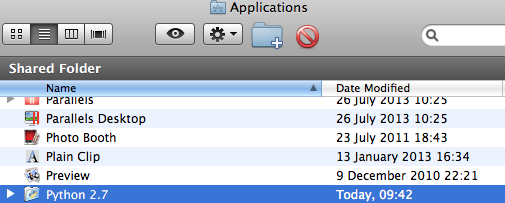
In the “Applications” folder, verify if Python 2.7 is already present.
Requirements: any operating system (Linux, Mac OS X, Windows, ...) with Python 2.7 and IDLE.
In the “Applications” folder, verify if Python 2.7 is already present.
If not, download Python 2.7 from http://www.python.org/downloads/.
(IDLE is included in this installer). Install the downloaded
version of Python as usual.
For example, for Mac users:
Double-click to open the downloaded “.dmg” file. The window represented in the following figure will open automatically: double-click on the “Python.mpkg” file to install Python.
For example, for Mac users:
Double-click to open the downloaded “.dmg” file. The window represented in the following figure will open automatically: double-click on the “Python.mpkg” file to install Python.
When the Python 2.7 appears in the “Applications” folder, the system is properly configured.
Section A - parse_entrezgene.py
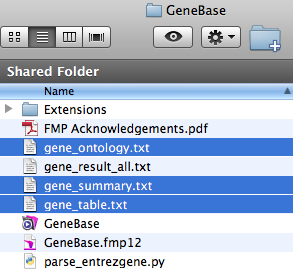
1. Go to the GeneBase folder.
2. If not already present, save a copy of the script file of interest in this folder.
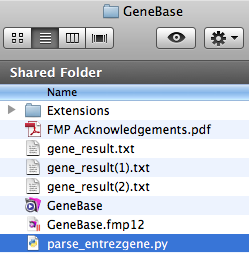
Please note that, in order to correctly
execute the script, you need to have the following files in
the same folder:
- parse_entrezgene.py;
- one or more files downloaded from the NCBI Gene website (usually automatically named gene_result.txt, gene_result(1).txt and so forth if more than one).
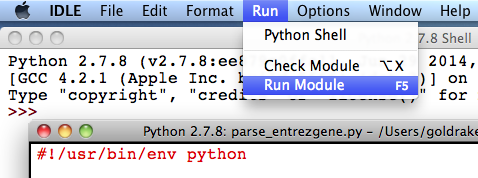
3. Double-click on the “parse_entrezgene.py” script file and two windows will appear: the “Python Shell” and “IDLE”.
4. To execute the script, keeping the “parse_entrezgene.py” window active, select “Run Module” from the “Run” menu.
- parse_entrezgene.py;
- one or more files downloaded from the NCBI Gene website (usually automatically named gene_result.txt, gene_result(1).txt and so forth if more than one).
3. Double-click on the “parse_entrezgene.py” script file and two windows will appear: the “Python Shell” and “IDLE”.
4. To execute the script, keeping the “parse_entrezgene.py” window active, select “Run Module” from the “Run” menu.
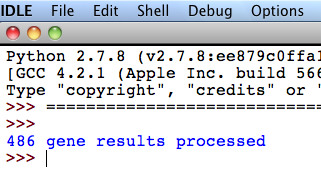
5. The programme is finished when the message “XXX gene results processed” appears in the “Python Shell”, where XXX is the number of NCBI's Gene entries downloaded in the first step of GeneBase tutorial.
In the GeneBase folder you will obtain three tab-delimited files: “gene_ontology.txt”, “gene_summary.txt” and “gene_table.txt”.
To import these files into GeneBase software, please go back to GeneBase tutorial step 1.3.
Section B - calculate_sequences.py
1. Go to the GeneBase folder.
2. If not already present, save a copy of the script file of interest in this folder.
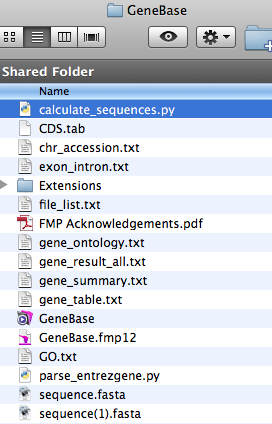
Please note that, in order to correctly
execute the script, you need to have the following files in
the same folder:
- calculate_sequences.py;
- exon_intron.txt;
- FASTA file(s) with the downloaded chromosome sequences, in this example sequence.fasta and
sequence(1).fasta;
- file_list.txt (with a list of FASTA file names).
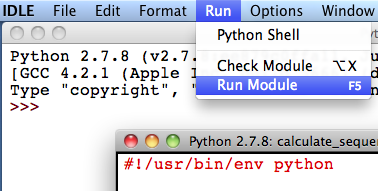
3. Double-click on the “calculate_sequences.py” script file and two windows will appear: the “Python Shell” and “IDLE”.
4. To execute the script, keeping the “calculate_sequences.py” window active, select “Run Module” from the “Run” menu.
- calculate_sequences.py;
- exon_intron.txt;
- FASTA file(s) with the downloaded chromosome sequences, in this example sequence.fasta and
sequence(1).fasta;
- file_list.txt (with a list of FASTA file names).
3. Double-click on the “calculate_sequences.py” script file and two windows will appear: the “Python Shell” and “IDLE”.
4. To execute the script, keeping the “calculate_sequences.py” window active, select “Run Module” from the “Run” menu.
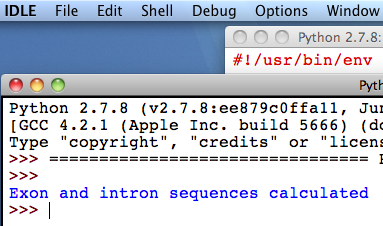
5. The programme is finished when the message
“Exon and intron sequences calculated” appears in the “Python
Shell” (this could take some hours).
In the GeneBase folder you will obtain a tab-delimited file named “exon_intron_seq.tab”.
To import this file into GeneBase software, please go back to GeneBase tutorial step 2.3.