INTRODUCTION
This online tutorial is designed to help first time GeneRecords
1.0 users.
It teachs how to install the software and how to import the desired
GenBank
entries in the database.
Please refer to the user Guide
for more detailed information.
Download GeneRecords 1.0 for
Mac OS 9 or Mac OS X from address:
http://apollo11.isto.unibo.it/software/GeneRecords_1.0/GeneRecordsMacOS9.sit
http://apollo11.isto.unibo.it/software/GeneRecords_1.0/GeneRecordsMacOSX.sit
The downloaded file should to be automatically decompressed,
generating a "GeneRecords" folder:
"GeneRecords 1.0 Mac OS 9" or "GeneRecords 1.0 Mac OS X" folder,
respectively.
If it does not happen, the decompression needs the "Stuffit Expander"utility.
The GeneRecords Folder contains:
GeneRecords file (runtime application),
42 related subdatabases (.DNA files),
the "SelfReplace GeneRecords Filter" helper application along with
its
"SelfReplace Pref" preference document,
the LocusLink database formatted for GeneRecords relationships.
The "Fasta extractor" is an utility to generate sequence in FASTA
format.
The Mac OS X version also includes the "Open Classic" application
(see below).
The "MacOS_Tutorial" and "MacOS_Guide" folders contain a copy
of the on-line documentation, for local (off-line)
use.
GeneRecords is based on FileMaker Pro 6 (FileMaker Pro, Inc.)
database management software (www.filemaker.com/index.html).
It is a set of related FileMaker Pro templates that will import
any set of
GenBank-formatted entries into a local database,
for retrieval and local elaboration of the text and sequence data.
INSTALLATION
Put the file "SelfReplace Pref" contained in the GeneRecords folder
into
the"Preferences" folder of the "System Folder".
Note:
Mac OS 9: "System Folder" is the name of the standard system folder
Mac OS X: "System Folder" is the name of the folder containing
Mac OS 9
"Classic"
compatibility environment.
"Classic"
is required for running GeneRecords,
check
that it is open by double-clicking on "Open Classic.app"
file
included in "GeneRecords folder").
USE
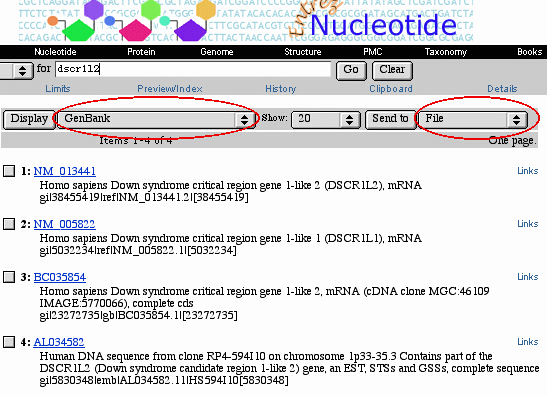
1. Download GenBank entries
Download a set of desired GenBank entries
(GenBank entry sequence allowed maximum size: up to 1,102,200 bp)
from any database which supports GenBank format
(GenBank "CON" division, i.e. NT_
entry code, is not supported).
There are two usual alternatives:
I. Querying GenBank via
World Wide Web at:
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Limits&db=Nucleotide
Users have to save the obtained entry set as follows:
choose "GenBank" format from the pop-up Menu
at the right of "Display" button,
choose "File"
from the pop-up Menu
at the right of "Send to" button,
then click on "Send to" button and choose "Save" button
in the next dialog box.
II. Performing download of large
data set via ftp at:
ftp://ftp.ncbi.nih.gov/genbank/
(decompress the files when appropriate)
At the end of this step, the users should have a text file
in GenBank flat file format,
containing the sequences to be imported into the GeneRecords database.
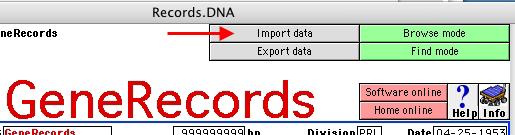
2. Import GenBank Entries
Open the "GeneRecords" file into the "GeneRecords 1.0" folder.
This action will open the file "Records.DNA", that represents the
"master" file of the program, linked and related to the
feature-specific subdatabases.
Click on the "Import data" button and follow instructions on the
dialog box
(choose the file to be imported)
This step may require a long time,
depending on the size of the original data file.
A set of 42 related files will be automatically updated with
the new data,
with each type of information imported in the appropriate file/field.
The actual number of imported records is shown in the left side
of the
Records.DNA window:
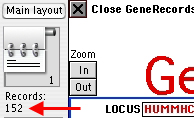
Note that each GenBank entry is imported as a single record.
You may adjust the layout appearance using "Zoom In"/"Zoom out"
buttons,
or clicking on the small resizing buttons at the bottom left corner
of any window.
The "Layout Menu" is a pop-up Menu in the
top left corner,
above the small book icon (see the Figure
above).
Within one database file, separate layouts may be provided.
Layouts determine how data is displayed.
Changes in a field present on a layout are reflected in the same
field
on all the layouts in the database.
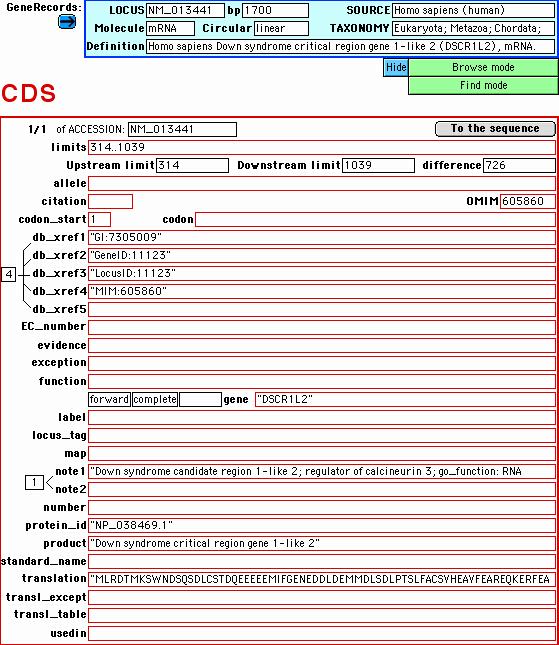
Each "feature" (e.g., exon) of the entry is visualized in a dedicated
subdatabase (e.g., "Exon.DNA" file),
while each field of the subdatabase corresponds to a "Feature Qualifier"
according to GenBank Format (e.g., "exon number").
--
You can move among different subdatabases,
clicking on the "To features" button,
![]()
then clicking on a single feature (green button).
You will be moved to the layout of the selected feature, e.g.: CDS:

Using the blue buttons,
you can quickly display in the respective, related subdatabase:
all the features occurrences in the current entry ("Cur." blue
button); or
all the feature occurrences in the entry set currently selected
("Found") in the master database ("Total" blue
button).
--
You may also directly choose a particular subdatabase
from the "Window" Menu, in the master "Records.DNA" file.
For example, this is the coding sequence (CDS) feature subdatabase,
fully searchable in each field:
--
When you visualize and search the content of each subdatabase
from within the master file,
you can switch among different features also by the "Layout Menu":
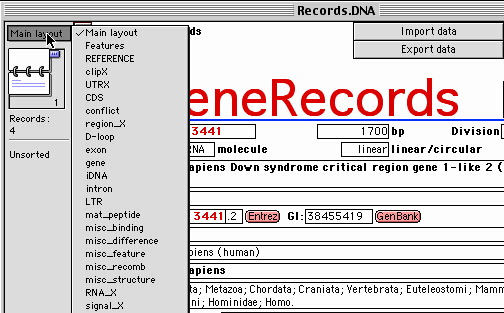
Please note that some features of the same type will be imported
in the same subdatabase; in particular:
the "ClipX" database includes the features: 5'clip, 3'clip;
the "UTRX" database includes the features: 5'UTR, 3'UTR;
the "Region_X" database includes the features: C_region, S_region,
V_region;
the "RNA_X" database includes the features:
misc_RNA, precursor_RNA, prim_transcript,
rRNA, scRNA, snRNA, snoRNA, tRNA;
the "Segment_X" database includes the features: V_segment, D_segment,
J_segment,
N_region;
the "Signal_X" database includes the features:
attenuator, CAAT_signal, enhancer, GC_signal,
misc_signal, promoter,
TATA_signal, terminator, -10_signal, -35_signal.
BROWSE MODE (NAVIGATION)
The FileMaker Pro based database may be used basically in these
"modes":
"Browse", "Find" and "Preview".
Switching among different modes can be made from the "View" Menu,
or from the respective green
"Browse mode""Find mode" buttons
available on the software windows.
In the "Browse" mode,
browsing the records set can be made clicking on the small book
icon
in the upper left corner:

In the GeneRecords database the users find four types of coloured button:

The RED buttons allow to open windows
of the default web browser,
to show the related site on the Internet:
![]()
The GREEN buttons allow shifting among
different layouts
(i.e., different visualization mode) of the same database file:
![]()
The GRAY buttons activate a predefinite instruction, e.g.:
![]()
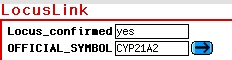
The BLUE buttons allow linking of the
related data
stored in the specific GeneRecords subdatabase:
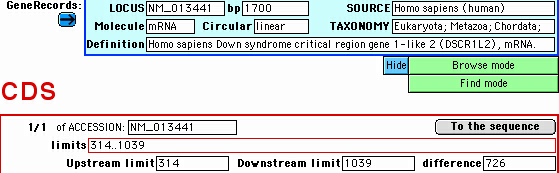
ACCESS TO SEQUENCE DATA
Nucleotide sequence of each feature each entry are provided
in the respective subdatabase.
Sequences are split into chunks of 50,100 bp.
To view the sequence of a desired feature,
click on "To the sequence" button in the respective subdatabase:
In some cases, the sequence is not immediately visualized
in the subdatabases.
To visualize the actual sequence chunk data
in the current record of subdatabase,
you should click on the button "Click to extract",
or choose the command "Extract this sequence" from the Menu "Actions".
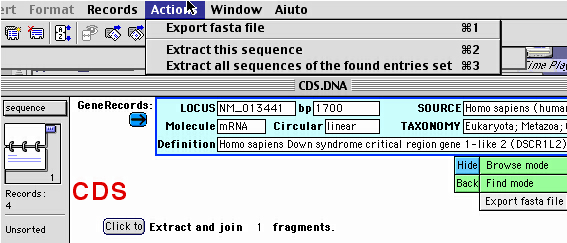
The command "Extract all sequences of the found entries set"
(from the Menu "Actions")
will extract the feature sequence for all the currently found records
set.
A FASTA format sequence export function is accessible in each
feature subdatabase, from the "Sequence" layout,
clicking on the "Export fasta file" button (see the Figure above).
SEARCH ("FIND") MODE
Switching among different modes can be made from the "View"
Menu,
or by clicking on the "Find mode"
green button in the database window.
In the "Find" mode, the small book icon in the upper left corner
represents different "requests" that are made for searching
in the database.
In the "Find" mode,
the user can fill a blank form allowing searching in specific
fields,
and by moving among the different layouts,
very complex searches can be made by
combining searches in different subdatabases
(each corresponding to a feature)
from within the master file "Records.DNA"
(which is dinamically related to the content of each subdatabase).
You can move among different layouts:
using the Layout (pop-up) Menu (Layout Menu),
clicking
on the bar at the top of the book icon; or:
using the green buttons available in the "Features" layout,
clicking
on the "To features" green button.
When searching in the master ("Records.DNA") database,
if one entry contains more recurrences of a feature,
all related records of the respective feature
subdatabase
are displayed in the master database corresponding layout.
In FileMaker Pro "Find" mode, the "AND" - "OR" - "NOT" operators
may
be used in a search in this way:
"AND" by filling in different fields located in the same "Request",
"OR" by generating additional requests
(from "Requests" Menu) in the same
query,
"NOT" by generating additional requests (from "Requests" Menu)
and checking the "Omit"
box.
The "Symbols" pop-up Menu in the "Find" mode allows query of
ranges, duplicates, wildcards and so on.
Each feature subdatabase can
be also individually searched,
after selection from Menu "Window".
The searching results are entries subsests matching the desired criteria.
The "Find record" script looks for a required corresponding record
group
(features) in the specific databases;
if one entry contains more recurrences of a feature,
all related records of the subdatabase are displayed.
---
Software limits:
Maximum size of GenBank entry to be imported: up to 1,102,200 bp.
Maximum size of GenBank ŇFeaturesÓ section for each entry,
allowing a correct Features splitting:
64,000 characters following text processing.
Entries with a larger ŇFeaturesÓ section will be processed,
but the splitting of the features in the subdatabases could be
incomplete.
A single GeneRecords database may store up to 2 Gbyte
(a physical limit of the core database).
The "CON" division of GenBank contains data for joining other sequences,
and it can not be imported.
Supplementary informations:
Detailed explanation of the GenBank Flat File format may be found
at:
ftp://ftp.ncbi.nih.gov/genbank/gbrel.txt
The definition and explanation of each GenBank Feature may be found
at:
http://www.ncbi.nlm.nih.gov/projects/collab/FT/index.html
General information about the Filemaker core functions may be found
at:
http://www.strath.ac.uk/CC/Courses/FilemakerPro/filemaker.html
http://www.wellesley.edu/Computing/Filemaker/filemaker4_tutorial.html
We enclose the LocusLink database, maintained at NCBI:
http://www.ncbi.nlm.nih.gov/LocusLink/
http://research.nhgri.nih.gov/microarray/downloadable_cdna.html
as an example of creating a relationship among different databases
using Generecords.
Bugs report:
Please report any bug or problem to:
pierluigi.strippoli@unibo.it
p.daddabbo@biologia.uniba.it