THE EXPRESSED SEQUENCE TAGS
(ESTs)
The method for rapidly obtaining
partial
mRNA sequences, published by
Adams et al. in
1991 and described below, gave a tremendous and sudden
acceleration to the rhythm of identification and cloning of mRNA,
to the point that during the 1990s the cloning in the form of cDNA
of about 1,000 new human mRNAs / year was reported in the
literature. In this way, the characterisation of the entire series
of the approximately 20,000 cDNAs corresponding to human RNAs
coding for proteins was substantially completed in the first years
after 2000.
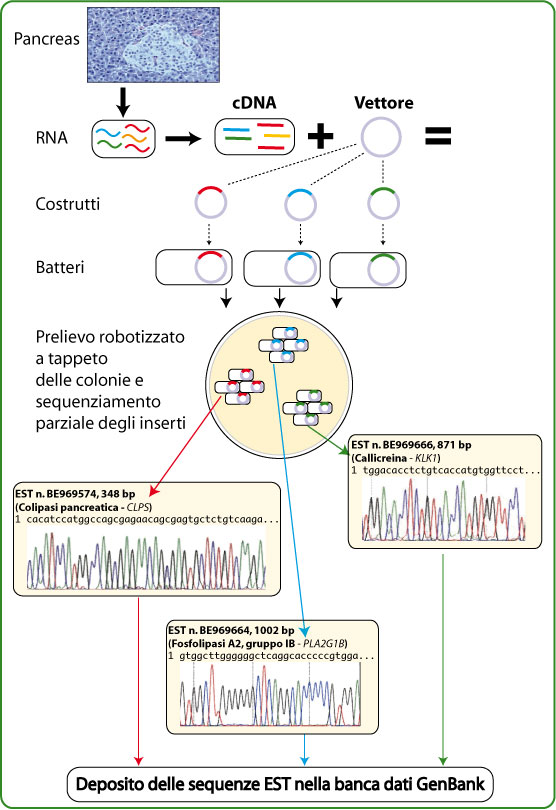
The fundamental idea to arrive at a faster
identification of the transcribed sequences was to create
databases containing partial sequences derived from RNA. To this
end, the
RNA extracted from various tissues is converted
into
cDNA with the use of reverse transcriptase and cloned
according to the cDNA library method. While until then the typical
approach aimed to seek only a specific sequence of interest,
present in a particular bacterial clone that had incorporated it,
the new strategy was distinguished by the intensive
automation
of the analysis procedure, which allowed to randomly characterise
a
large number of bacterial clones, favouring speed over
accuracy and completeness of sequencing (
Figure,
from Fantoni et al., "Genetica", Piccin).
In this way, it is possible to determine the sequence of
a few
hundred bases located at one of the two ends of the cDNA insert,
which on the whole is usually about 1-3 kb long. The sequences
obtained in this way are called
EST (expressed sequence tags),
that is, partial RNA sequences. A
single EST
is, therefore, a short sequence located at one end of a specific
cDNA, or more briefly represents a fragment of a specific RNA.
ESTs are published in specialised databases (Boguski et al., 1993)
and are publicly available for computer sequence analysis.
Furthermore, representative cells of each bacterial colony are set
aside by the robots and stored at low temperature to allow the
replication of the cloned cDNA as desired and its complete
characterisation, if its sequence is of interest.
It is therefore assumed by definition that, if a particular
sequence of bases is found in the EST database, it belongs to a
transcript. It is also possible to "assemble" by the
computer the fragments of EST sequences referable to the same
transcript by exploiting the existence of regions of coincident
sequence in the various fragments obtained at random. In this way,
it is possible to quickly identify, by only computer processing,
genome sequences that have been transcribed (genes) without having
to deal with the "background" of extragenic sequences, as happens
in traditional "genome projects".
Since different RNAs are present in the cells
depending on the tissue to which they belong and the conditions in
which they are found, dozens of cDNA libraries are prepared in the
larger EST projects, each starting from RNA extracted from a specific
tissue or organ which is in a given physiological (e.g.,
stage of development) or pathological (e.g., neoplastic
proliferation) condition.
The availability of the complete sequence of the human genome and
the development of powerful bioinformatic analysis tools make it
possible today to determine the position of the EST on the
chromosome with the simple comparison conducted on the computer
between an EST sequence and the complete sequence genomic DNA.
{kind=link}